Die
Arrhythmogene Rechtsventrikuläre Kardiomyopathie (AVRCM)
Amerikanisch
auch Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy- ARVD
Die arrhythmogene, rechtsventrikuläre Kardiomyopathie ist eine seltene Erkrankung der Herzmuskulatur die überwiegend auf einer angeborenen Störung des Aufbaues der Herzmuskelzellen beruht. Sie tritt vermehrt bei Männern im Alter zwischen 20 und 40 Jahren auf. Ihr Kennzeichen ist die Umwandlung der Muskulatur der freien Wand der rechten Herzkammer in Fettgewebe. Als Folge entstehen umschriebene Aussackungen der Herzwand (Dysplasien oder Aneurysmata genannt). Familiäre Häufungen können auftreten.
Die Hauptsymptome sind Herzstolpern infolge eines unregelmäßigen Herzschlags (Herzrhythmusstörungen durch so genannte ventrikuläre Extrasystolen) oder seltener Schwächeanfälle und Ohnmachten. Das Auftreten eines plötzlichen Herztodes durch bösartige Herzrhythmusstörungen aus der rechten Herzkammer wird in Einzelfällen beobachtet, ist allerdings selten.
So dramatisch dies auch klingt, in der Regel führen die meisten Menschen, die unter einer ARVCM leiden ein ganz normales Leben mit gelegentlichen Episoden von Herzstolpern oder Herzrasen. Die betroffenen Patienten benötigen, wenn überhaupt, nur wenige, in der Regel gut verträgliche Medikamente. In Einzelfällen kann die Erkrankung jedoch auch gravierende Folgen in Form lebensgefährlicher Herzrhythmusstörungen oder einer Herzschwäche haben. Daher sollte die Untersuchung und ggf. auch die Behandlung in den Händen von Kardiologen liegen, die mit dieser Erkrankung besonders vertraut sind. Es ist wichtig zu wissen, dass bei der ARVCM lediglich die Beschwerden, also in erster Linie die Herzrhythmusstörungen, behandelt werden. Eine Möglichkeit zur Beseitigung der Grundkrankheit selbst ist bisher nicht bekannt. In seltenen Einzelfällen kann die Erkrankung so schwer verlaufen, dass sogar eine Herztransplantation erwogen werden muss. Bei schwerwiegenden Herzrhythmusstörungen kann ein implantierbarer Spezialschrittmacher mit Defibrillator (ICD) notwendig werden.
Es handelt sich bei der ARVCM um ein vielschichtiges Krankheitsbild, das für den Betroffenen und häufig auch die behandelnden Hausärzte anfangs schwer zu verstehen ist. Das Ziel der folgenden Darstellung ist es, die wesentlichen Informationen zu geben, die für den täglichen Umgang mit der Erkrankung und die weitere Lebensplanung hilfreich sind. Um dieses Ziel zu erreichen, ist es an einigen Stellen notwendig, spezielle Themen ausführlich darzustellen. Diese Informationen sind für Patienten und Angehörige gedacht. Sie können aber auch interessierten Ärzten, die nicht regelmäßig mit diesem Krankheitsbild umgehen, wichtige Hinweise geben.
Der Name der Erkrankung Arrhythmogene Rechtsventrikuläre Cardiomyopathie, abgekürzt ARVCM, leitet sich so ab: Arrhythmogen = Herzrhythmusstörungen hervorrufend, Rechtsventrikulär = die rechte Herzkammer betreffend, Cardiomyopathie (im Deutschen heute Kardiomyopathie) = Herzmuskelerkrankung (Kardio = Herz, myo = Muskulatur, pathie = Krankheit).
Um das Folgende gut verstehen zu können, ist es gut, sich noch einmal an die Funktion des Herzens zu erinnern. Beginnen wir mit der rechten Herzkammer (rechter Ventrikel), die hauptsächlich von der ARVCM befallen wird. Diese Kammer saugt das sauerstoffarme, vom Körper kommende Blut aus den Körpervenen über die rechte Vorkammer an und pumpt es in die Lunge weiter. Dort wird das Blut von der im Körper entstandenen Kohlensäure (CO2) befreit und wieder mit Sauerstoff auf 100% gesättigt. Die linke Herzkammer saugt nun das sauerstoffreiche Blut aus der Lunge über die linke Vorkammer an und pumpt es mit hohem Druck – dem Blutdruck – über die Schlagadern weiter in den Körper. Die notwendige Pumpkraft wird durch die Herzmuskelzellen erzeugt, aus denen die Wand der Herzkammern zum weit überwiegenden Teil besteht. Der übrige Teil der Herzwand wird durch Bindegewebszellen, Adern und einzelne Fettzellen gebildet. Im Aufbau der Herzkammern unterscheidet man die freie Kammerwand von der Herzscheidewand (Ventrikelseptum).
Krankheitsfolgen am Herzmuskel
Bei der ARVCM kommt es in umschriebenen, fleckförmigen Gebieten, aber auch diffus verteilt in der freien rechten Herzkammerwand zu einer schweren Schädigung einzelner Herzmuskelzellen. Diese Herzmuskelzellen sterben vorzeitig ab und werden Schritt für Schritt in Fettgewebszellen umgewandelt. Die Herzwand „verfettet“. Dieser Prozess führt meist zur Ausbildung von umschriebenen Aussackungen der Herzwand, die „Dysplasien“ oder „Aneurysmata“ genannt werden. Aber auch Formen mit einer diffusen, flächigen Ausbreitung der Erkrankung sind bekannt. Ein Mitbefall der linken Herzkammer wird insbesondere bei fortgeschrittener Erkrankung in bis zu 50% der Fälle beobachtet. Dieser Prozess hat zum einen die zunehmende Schwächung der Pumpkraft des rechten Herzens und zum anderen das Auftreten einer verlangsamten Leitung des Herzstroms in den verfetteten Gebieten zur Folge, was zum Auftreten eines unregelmäßigen Herzschlages oder von Herzrasen führen kann.
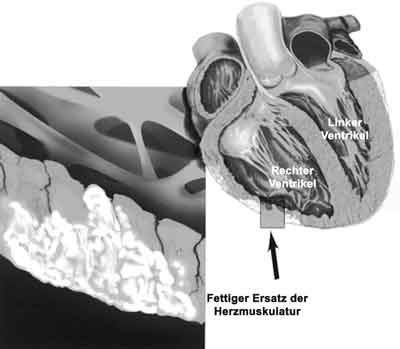
Abbildung 1: ARVCM, mit Erkrankung der Wand des rechten Ventrikels
Die durch die Verfettung entstandene, ungleichmäßige Zusammensetzung der Herzwand führt auch zu einer ungleichmäßigen, damit gestörten Ausbreitung der elektrischen Ströme, die die Herzmuskelzellen zum Pumpen anregen. Die Folge einer solchen Störung ist das Auftreten von zusätzlichen, Herzschlägen, so genannten „Extrasystolen“ die zu einem unregelmäßigen Herzschlag führen. Bei der ARVCM liegt der Ursprung dieser „Herzrhythmusstörungen“ daher typischerweise in der rechten Herzkammer. Extrasystolen werden von einem Teil der Menschen als „Herzstolpern“ verspürt, eine große Zahl der Menschen bemerken sie jedoch gar nicht. Treten solche irregulären Herzschläge stark gehäuft und in rascher Folge auf, so kann es zu einer so starken Beschleunigung des Herzschlages kommen, dass die Herzkammern nicht mehr genug Zeit haben sich zu füllen und zu entleeren (Herzrasen). Dies nennt man auch „Kammertachykardie (ventrikuläre Tachykardie). Meist dauert diese jedoch nur wenige Sekunden und geht von selbst wieder in einen normalen Herzschlag über.
Hält die Tachykardie jedoch länger an, so kommt es zu Schwindel und Schwächeanfällen, weil die Durchblutung des Gehirns nicht mehr ausreicht. Nur bei sehr schnellen Kammertachykardien kann es zu Ohnmachten (Synkopen) kommen. Hält die Kammertachykardie über eine längere Zeit an, dann kann sie in „Kammerflimmern“ übergehen. Das Kammerflimmern kann jedoch auch plötzlich, ohne eine vorangegangene Kammertachykardie auftreten. Im Zustand des Kammerflimmerns kann das Herz nicht mehr pumpen und hat auch ohne äußeren Eingriff durch einen geschulten und entsprechend ausgerüsteten Helfer oder Arzt keine Möglichkeit, sich wieder zu stabilisieren. Durch die Herzmuskelverfettung, die durch die ARVCM entstanden ist, kommt es zur Verminderung der Pumpkraft des Herzens. Diese Verminderung wird anfangs erfolgreich durch eine Vergrößerung der Herzkammer und eine als Reaktion auf die Mehrbelastung auftretende Verdickung der verbliebenen Herzmuskelzellen ausgeglichen. Sobald diese Reserven jedoch aufgebraucht sind, kommt es zur Ausbildung von Zeichen der Rechtsherzschwäche. Diese macht sich in Form eines Rückstaus des Blutes in die Körpervenen bemerkbar. Die Halsvenen sind auch im Sitzen verdickt zu sehen, die Leber schwillt schmerzhaft an und in den Beinen lagert sich Wasser ein (Ödembildung). Dieser Prozess setzt schleichend ohne Schmerzen, teilweise im Laufe vieler Jahre ein und wird daher häufig auch erst spät bemerkt. In Einzelfällen kommt es, besonders in höherem Lebensalter und bei langem Krankheitsverlauf, auch zu einer bedeutsamen Minderung der Pumpkraft der linken Herzkammer.
Genetische Grundlagen
Die Krankheit tritt familiär gehäuft auf. Als Ursache für diese Erkrankung sind bei 30 bis 50% der Erkrankten Störungen im Erbgut festgestellt worden. Es handelt sich bei ca.30% bis 50% der Erkrankungen um eine autosomal dominant vererbte 'Form. Männer sind häufiger befallen als Frauen (Verhältnis 1,6:1). Wenn ein Elternteil ein krankhaft verändertes Gen besitzt, hat jedes Kind eine 50% Wahrscheinlichkeit, die Erbanlage für diese Erkrankung zu erhalten. Bisher sind auf mehreren Chromosomen und unterschiedlichen Genabschnitten typische Veränderungen für die ARVCM aufgedeckt worden. Durch den fehlerhaften Aufbau einzelner Abschnitte der Gene ist der Aufbau der Verbindung zwischen den Herzmuskelzellen (Interscalated discs mit Desmosomen, Gap Junctions und fascia adherence) gestört. Kommt es zu wegen dieser Störung zu einer Ablösung der Herzmuskelzelle aus dem Zellverband, so wird diese absterben und durch Fettgewebe ersetzt. Dieser Verlust der Verbindung zwischen den Herzmuskelzellen wird möglicherweise durch körperliche Belastungen gefördert. Es wird unter Wissenschaftlern diskutiert, ob aus diesem Grund diese Erkrankung bei sportlich aktiven Menschen gehäuft in besonders ausgeprägter Form beobachtet wird. Die Erkrankung wird selten vor der Pubertät und meistens zwischen dem zwanzigsten und vierzigsten Lebensjahr festgestellt. Die ARVCM ist eine seltene Krankheit. Die Schätzung ihrer Häufigkeit ist wegen der hohen Dunkelziffer und der Ungenauigkeit der wissenschaftlichen Untersuchungsergebnisse weitgehend auf Spekulationen angewiesen. Neu beobachtete Erkrankungen werden bei bis zu 0,06% der Bevölkerung (d.h. 1 von 60.000 Einwohnern) pro Jahr angegeben. Jedoch bestehen starke regionale Unterschiede, selbst innerhalb einzelner Länder.
Wie wird die ARVCM festgestellt?
Symptome
Untersuchungsverfahren
Die körperliche Untersuchung ergibt, insbesondere im frühen Stadium, keine verwertbaren Hinweise auf die Erkrankung. Es gibt keinen einfachen apparativen Test oder eine einzelne Untersuchungsmethode, um eine ARVCM sicher festzustellen oder auszuschließen. Daher ist es im Verdachtsfall notwenig, eine gründliche Untersuchung mit Einschluss mehrerer Untersuchungsverfahren bis hin zur Herzkatheteruntersuchung durchzuführen.
Der relativ große Untersuchungsaufwand ist jedoch gerechtfertigt, da die Sicherung der Diagnose oder ihr Ausschluss für Menschen mit Beschwerden in Form von Herzstolpern, Herzrasen, Ohnmachten oder plötzlichen Herztoden in der Familie, und speziell für Hochleistungssportler von großer Bedeutung ist. Nur im Falle einer möglichst klaren Diagnose kann eine frühzeitige Behandlung beginnen und die bestehenden Risiken können so weit wie möglich minimiert werden.
Ruhe-EKG
Besonders
wichtige Hinweise gibt das EKG in Ruhe. Typische
EKG-Zeichen sind:
Negative, nach unten weisende T-Zacken über den rechtspräkordialen Ableitungen
V1+ V2, in schweren Fällen der Nachweis einer Verbreiterung des QRS –Komplexes
> 110ms oder die Ausbildung einer Epsilon Welle in V1-V3, und darüber
hinaus auch eine längere QRS-Dauer V1+V2+V3 im Vergleich zu V4+V5+V6.
(Das Verhältnis der QRS-Dauer in V1+V2+V3/ V4+V5+V6 >1,2).
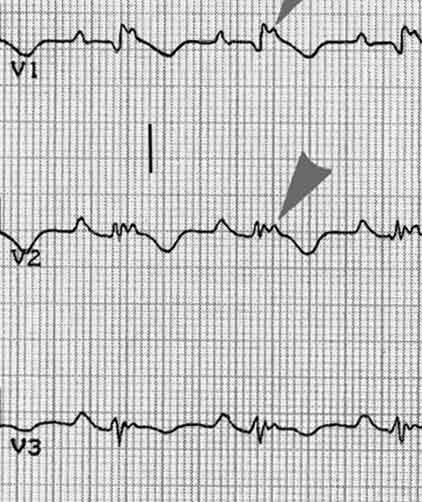 |
Abbildung
2: Epsilon Welle und negative ST-Zacken
(Quelle Braunwald, Heart Disease, 6th Ed. Saunders)
Wird das EKG während einer Phase mit Herzstolpern (Extrasystolen) oder mit schnellem Herzschlag (Tachykardie) aufgezeichnet, so erkennt man, dass die Rhythmusstörung ihren Ursprung in der rechten Herzkammer hat. Die Extrasystolen oder die Tachykardien zeigen die für die ARVCM typische Form eines „Linksschenkelblocks". Wenn zum Zeitpunkt des Auftretens von Herzrhythmusstörungen ein EKG aufgezeichnet wird, kann der Ursprung der Herzrhythmusstörungen genau analysiert werden.
Zur Aufdeckung der Art und der Häufigkeit der Herzrhythmusstörungen werden weitere EKG-Verfahren eingesetzt.
Eine Zunahme der Extraschläge oder das Auftreten von Herzrasen unter körperlicher Belastung sind typisch für eine ARVCM. Daher ist es wichtig, dass regelmäßig auch ein Belastungs-EKG durchgeführt wird.
Für die Feststellung, die Beurteilung von Ausmaß und den Verlauf einer ARVCM ist die Häufigkeit und die Schwere der Rhythmusstörungen wichtig. Die Kenntnis der Zunahme einer Häufung von Rhythmusstörungen in Ruhe oder unter Belastung hilft bei der Therapieentscheidung und Überprüfung der Wirksamkeit einer Therapie.
Signalmittellungs-EKG (Spätpotential-EKG /Late-Potential )
Echokardiografie
Eine weitere, moderne Methode stellt die Kernspintomografie, auch Magnetresonanz-tomografie (MRT) genannt, dar. Diese Methode macht ohne Röntgenstrahlen, allein auf dem Boden von starker Magnetisierung der Körpermoleküle Schnittbilder durch den menschlichen Körper und damit auch durch das Herz.
Es sind nicht nur Bewegungsstörungen der Herzwand, sondern auch feine Veränderungen in der Zusammensetzung der Herzmuskulatur, speziell auch Fetteinlagerungen zu sehen. Von den bildgebenden Verfahren ist die Magnetresonanztomografie bereits heute die sensitivste Methode und übertrifft die Genauigkeit der Echokardiografie. Jedoch ist sie in Einzelfällen nicht ausreichend empfindlich, die Diagnose ARVCM zu sichern. Diese Untersuchungen des Herzens gelingen nur mit kostspieligen und speziell für Kardiologen ausgerüsteten Geräten. Die Erstellung und Beurteilung der Bilder bedarf einer speziellen Ausbildung.
Weitere Sicherheit erhält man erst im Rahmen einer Herzkatheteruntersuchung. Bei der Darstellung der rechten und linken Herzkammer mit Röntgenkontrastmittel kann man in der Regel auch umschriebene Pumpstörungen und die Auswirkung der Erkrankung auf die Pumpkraft des Herzens erkennen. Mittels einer kleinen, an der Spitze eines Herzkatheters befestigten flexiblen Biopsiezange kann man nahezu schmerzfrei stecknadelkopfgroße Herzmuskelproben aus der Wand der rechten Herzkammer entnehmen, die man unter dem Hochleistungsmikroskop und mithilfe von Fettgewebsfärbungen auf den Fettgewebsanteil des Herzmuskels hin untersuchen kann. Dabei muss man sich jedoch bewusst sein, dass ein Ausschluss oder der Beweis einer ARVCM mittels einer solchen Herzmuskelprobe nicht möglich ist, da die Krankheit häufig nur fleckförmig und meist nicht in der Herscheidewand auftritt, an der aus Sicherheitsgründen in der Regel die Gewebsproben entnommen werden. Aus diesem Grunde ist die Herzmuskelbiopsie nur in wenigen Fällen angezeigt.
|
|
|
Systole |
Diastole |
| Die
weissen Pfeile zeigen eine Dysplasie |
|
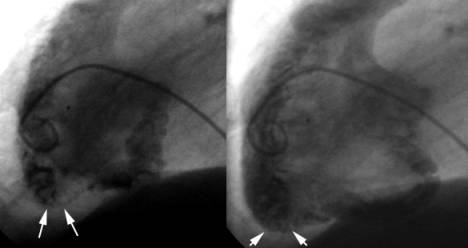
Abbildung 3: Rechte Herzkammer im Röntgenbild bei der Herzkatheteruntersuchung
Elektrophysiologische Herzkatheteruntersuchung (EPU)
Bei einer elektrophysiologischen Untersuchung wird über drei Ekg-Kabel (spezielle Herzkatheter), die mithilfe einer Röntgendurchleuchtung im Inneren des Herzens platziert werden, die elektrische Erregungsausbreitung über die Herzmuskelzellen vermessen. Zusätzlich werden die Rhythmusstörungen, die den Patienten im Alltag belasten, zur genauen Untersuchung gezielt mithilfe dieser Elektrodenkatheter ausgelöst. Die durch die Veränderungen des Wandaufbaus (Ersatz von Herzmuskelzellen durch Fettgewebszellen) und entstehenden Störungen der elektrischen Erregungsausbreitung lassen sich mit dieser Kathetertechnik noch genauer aufdecken als dies durch das Signalmittelungs-Ekg möglich ist. Auch kann diese Methode zuverlässig harmlose von gefährlichen Rhythmusstörungen unterscheiden. Die elektrophysiologische Herzkatheteruntersuchung ist ein sicheres und weitgehend schmerzfreies, jedoch mit Rötengenstrahlung verbundenes, relativ zeit- und personalaufwendiges Verfahren. Es wird daher nur bei Patienten mit bereits bestehendem Verdacht auf eine ARVCM und bedrohlichen Herzrhythmusstörungen eingesetzt. Kandidaten sind insbesondere Patienten mit Bewusstlosigkeiten, Kammertachykardien oder überlebter Wiederbelebungsmaßnamen (das Letztere wird auch häufig etwas irreführend als überlebter plötzlicher Herztod bezeichnet).
Eine sichere Feststellung oder ein Ausschluss einer ARVCM mithilfe einer Genanalyse ist bisher ebenso wenig möglich wie eine Vorhersage des weiteren Krankheitsverlaufes. Dies liegt an der verwirrenden Vielzahl der gefundenen genetischen Veränderungen. In schwerwiegenden Einzelfällen kann jedoch eine genetische Beratung durch eine geeignete Beratungsstelle, z. B. bei der Geburtenplanung sinnvoll sein. Die stürmischen Entwicklungen in der Genforschung lassen in näherer Zukunft eine deutliche Erweiterung der Untersuchungsmöglichkeiten auf diesem Gebiet erwarten.
Man muss sich bewusst sein, dass bei der überwiegenden Zahl der Erkrankten die richtige Diagnose nicht erkannt wird. Die ARVCM kann sehr lange stabil und ohne schwerwiegende Symptome bleiben. Wie häufig und wann ein Übergang in eine der schweren Formen eintritt, ist wegen der Schwierigkeiten der Erkennung der Krankheit und der Beurteilung ihres Schwergrades bisher unbekannt. Für den einzelnen Patienten lässt sich daher, wie oben bereits gesagt, der Verlauf der Erkrankung daher nicht vorhersagen.
Diese Überlegungen zeigen, wie wichtig es bei dieser seltenen Krankheit ist, schon bei dem Verdacht auf das Vorliegen einer ARVCM die Untersuchungsmöglichkeiten eines kardiologischen Zentrums in Anspruch zu nehmen. Der Vorteil liegt darin, dass hier regelmäßig Patienten mit dieser Erkrankung betreut werden. Dort besteht sehr viel eher die Möglichkeit, die endgültige Diagnose frühzeitig zu stellen und den richtige Behandlungsweg zu finden.
Die durch die Herzrhythmusstörungen ausgelösten Symptome der ARVCM können in der Regel gut behandelt werden. Jedoch ist bisher keine Behandlung bekannt geworden, die die Erkrankung der Herzmuskulatur selbst, das Absterben der Herzmuskelzellen und ihren Ersatz durch Fettgewebe, gezielt günstig beeinflusst oder verhindert. Auch das Nachwachsen von Herzmuskelzellen kann bisher nicht erreicht werden.
Herzrhythmusstörungen, die keine Beschwerden bereiten, erfordern keine Behandlung. Die meisten Menschen, die unter einer ARVCM leiden, führen ein ganz normales Leben mit gelegentlichen Episoden von Herzstolpern oder beschleunigtem Herzschlag. Es wird angenommen, dass eine starke körperliche Belastung die schwerwiegenden Herzrhyth-musstörungen besonders häufig hervorrufen kann. Daher sollten starke körperliche Belastungen, insbesondere Hochleistungssport, von Erkrankten vermieden werden. Diese Vorsichtsmaßnahme wird durch die Erfahrung begründet, dass die ARVCM für eine große Zahl der plötzlichen Herztode bei Männern im Alter unter 35 Jahren verantwortlich gemacht werden muss, die im Zusammenhang mit sportlichen Aktivitäten aufgetreten sind (z. B. Fußball, Marathonlauf). Als Medikamente zur Behandlung von Symptomen sind die Beta-Blocker besonders geeignet. Sie beschützen das Herz vor den Stresshormonen, die gerade unter körperlicher Belastung freigesetzt werden (z. B. Adrenalin). In besonderen Einzelfällen werden auch antiarrhythmische Substanzen wie Sotalol oder Amiodarone trotz eines relativ hohen Nebenwirkungsrisikos mit Erfolg eingesetzt.
Wenn die Herzrhythmusstörungen in Form von Herzrasen Beschwerden bereiten und Medikamente nicht ausreichen, kann eine Katheterablation häufig sinnvoll sein. Mithilfe von Spezialkathetern (Ablationskathetern) wird die Stelle in der rechten Herzkammer aufgesucht, an der die Rhythmusstörungen entstehen. Gelingt dies, kann durch ein Erwärmen der Katheterspitze mit Hochfrequenzstrom diese Stelle im Herzmuskel verödet werden. Viele Patienten sind nach einer solchen Behandlung beschwerdefrei. Da es sich jedoch in der Regel um eine weiter fortschreitende Krankheit handelt, ist dier Behandlungserfolg meist nicht von Dauer.
Allerdings verhindert diese Behandlung nicht, dass mit dem Fortschreiten der Erkrankung innerhalb des Herzmuskels andere Stellen entstehen, die erneut Rhythmusstörungen verursachen.
Für Patienten, die schwerwiegende Herzrhythmusstörungen wie Kammerflimmern überlebt haben, gibt es heute eine sehr wirksame Behandlungsmethode. Diese kann ebenfalls angewendet werden, wenn oben beschriebenen Kammertachykardien trotz anderer Behandlungsformen vermehrt auftreten, beziehungsweise so schnell sind, dass sie vom Patienten nicht ertragen werden können. Diese Behandlungsform kann auch vorsorglich für Menschen erwogen werden, in deren Familie viele unklare plötzliche Todesfälle im jungen Lebensalter vorgekommen sind. Da es sich um eine Behandlung handelt, die zum Glück selten erforderlich wird, dann jedoch von großer Bedeutung ist, soll ihre Funktionsweise im Folgenden ausführlicher dargestellt werden.
Die ICD Behandlung besteht in der Einpflanzung (Implantation) einer besonderen Art von Herzschrittmachern, die „Implantierbarer Defibrillator“ genannt werden (international als „ICD“ = „implanted cardioverter defibrillator“ abgekürzt). Dieser Schrittmacher wird unter die Haut unterhalb des linken Schlüsselbeines eingesetzt. Dort ist er vor Schädigungen geschützt und wasserdicht verpackt. Der ICD ist etwa so groß wie eine Streichholzschachtel, sodass es zu keiner bedeutsamen körperlichen Entstellung kommt.
Vom Defibrillator ausgehend, wird über die Blutgefäße, die zum Herzen führen (Venen), eine weiche, blutfreundliche Elektrode in die rechte Herzkammer des Patienten vorgeschoben und dort sicher verankert. Diese tastet die elektrischen Impulse des Herzens ab und leitet sie dem Defibrillator zur Begutachtung zu. Werden Herzrhythmusstörungen entdeckt, so überträgt die Elektrode die Behandlungsimpulse des Defibrillators auf den Herzmuskel. Die Implantation eines Defibrillators ermöglicht es, dass der Herzrhythmus 24 Stunden am Tag überwacht wird.
Durch die Programmierung des Gerätes wird erreicht, dass lebensbedrohliche Herzrhythmusstörungen selbstständig erkannt und mit gezielten elektrischen Impulsen oder Elektroschocks beendet werden. Die gezielten elektrischen Impulse zur Überstimulation der Herzrhythmusstörung werden vom Patienten nicht bemerkt. Gelingt die Überstimulation nicht oder ist das lebensgefährliche Kammerflimmern eingetreten, so wird dieses durch den Schrittmacher automatisch mit einem Elektroschock durchbrochen und rasch der normale Herzschlag wieder hergestellt. Dieses Verfahren ist zurzeit die sicherste Behandlung für lebensgefährliche Herzrhythmusstörungen. Man kann sich mit einem implantierten Defibrillator ungezwungen bewegen, duschen und auch sonst die üblichen alltäglichen Dinge ohne Sorge tun. Auch Auto fahren ist 6 Monate nach Implantation grundsätzlich möglich, solange kein Schock vom IDC ausgelöst worden ist. Wenn innerhalb von 3 Monaten nach Implantation kein Schock aufgetreten ist, kann das Autofahren sogar bereits nach diesen ersten 3 Monaten wieder aufgenommen werden. Dies gilt nicht, wenn andere Krankheitssymptome vorliegen.
Regelmäßige Kontrollen der Funktion des ICD-Gerätes führen die implantierenden Kliniken und auch niedergelassene Kardiologen in ihrer Praxis durch. Kontrollintervalle sind 3-6 Monate.
|
|
Abbildung 4: An dieser Stelle wird der ICD in der Regel implantiert.
Im Falle eines sehr schweren Verlaufes der Erkrankung mit Ausbildung einer hochgradigen Pumpschwäche des Herzens oder nicht beherrschbaren lebensbedrohlichen Herzrhythmusstörungen muss die Vorbereitung für eine Herztransplantation bei den in der Regel relativ jungen Patienten rechtzeitig in den Behandlungsplan mit aufgenommen werden.
Autor: Dr. Christian Leuner
Diese Literatur liegt dem Artikel zu Grunde
In Deutscher
Sprache:
In Englischer Sprache:
Arrhythmogenic right ventricular Dysplasia.Chalkins H, Markus F.,In Braunwald, E., ed. Harrison’s Advances in Cardiology. New York, NY: mc Graw-Hill 2003:378 – 383
Arrhythmogenic right ventricular cardiomyopathy/dysplasie: a not so rare "disease of the desmosome" with multiple clinical presentations. Herren, TH, Gerber P.A., Duru, F., Clin Rres Cardiol 2009;98:141-158
Im Internet erreichbar:
(besonders gut in englischer Sprache)
Cardiology Patient Page: Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
Circulation
2003;107:e31-e33. doi:10.1161/01.CIR.0000053943.38763.70-Cardiology Patient Page
ARVD/C Questions and Answers
John Hopkins University - Heart&Vascular Institut. Baltimore, Maryland, USA
http://www.hopkinsmedicine.org/heart_vascular_institute/clinical_services/centers_excellence/arvd/patient_resources/questions.html
Arrhythmogenic right ventricular dyplasia
An article from the ESC Council for Cardiology Practice
Fernández-Armenta J., Brugada J.
http://www.escardio.org/communities/councils/ccp/e-journal/volume10/Pages/management-ventricular-tachycardia-in-ARVD-Brugada-Josep.aspx#.Us_H2XmE6l4